Discuss on reddit.
Sassy is a tool for quickly searching short DNA patterns across large inputs that Rick Beeloo and myself have been working on over the past year. It just got published (Beeloo and Groot Koerkamp 2026b) (PDF, DOI), and this blog post briefly outlines some possible applications and how we developed it. See also the GitHub readme and the slides for my RECOMB-Seq talk, which loosely form the basis for this post.
Tl;dr:
Sassy takes a pattern, and then fuzzy-searches the input files, allowing some errors. It’s great for quickly checking your data for contamination from barcodes, adapters, primers, and so on, as well as for off-target detection.
It comes as a CLI, as well as Rust library with Python, C, and R bindings.
Sassy was developed for searching fasta and fastq DNA files, but also provides a
mode for searching plain ASCII files.
Below, I search a random pattern in a full human genome.
For \(k=2\) edits, there are no matches.
For \(k=3\) errors, there is one match with 2 substitutions (orange), and a deletion:
the red G that is in the pattern but not the text.
For \(k=4\), there are 19 additional matches.
For each match, it shows the filename that matches and the name of the record (chromosome, in this case). Then, it shows whether the match was on the forward (+) or reverse-complement (-) text, the number of errors, and a snippet of the record. This makes it especially easy to see whether matches are close to the start/end of a record.
Note that this video is real-time: searching a human genome both in forward and reverse-complement direction only takes around 2 seconds!
1 Why should you care? Link to heading
Before going into the technical details, let me (try to) convince you why you should care about Sassy.
Many sequencing techniques require some form of preparation of the sample where various types of artificial DNA are attached to all sequences before they are processed. For example:
- barcodes and adapters used for demultiplexing or other custom kits,
- tags and UMIs for single-cell RNA sequencing, and
- primers for sequencing viruses.
Before processing such data, one should make sure that either these signals are present at the start/end of each read, in case they are expected, or that they have been successfully stripped/trimmed in case they are not expected.
The Sassy CLI provides a quick way to check that the data looks as expected: you simply give it the pattern, a small threshold on the number of errors you allow (usually around 3 or so for patterns of length ~20), and then pass it the data files. It then prints all regions where the pattern matches, and you can quickly inspect whether this is at the expected start/end of each read.
While surely there are other tools that do something similar, I am not aware of anything that is both as fast and as simple to use as Sassy.
2 Using Sassy Link to heading
Since this post is aimed at potential users, let’s see how you can install and use Sassy before scaring everybody away with algorithms. For more details, see the GitHub readme.
You can either download the latest GitHub release, use
conda install -c bioconda sassy, or use cargo binstall sassy.
Precompiled binaries for x86-64 come with both AVX2 and AVX-512
support.1
2.1 The CLI Link to heading
The CLI provides a few subcommands: sassy grep, sassy search, and sassy filter
that all do the same, but output different results.
grep shows a grep-like human readable output,
search outputs a .tsv with search results, and
filter prints the (non) matching records.
Lastly, sassy crispr is specifically designed for Crispr off-target detection.
sassy agrep can be used to fuzzy search plain ASCII files:
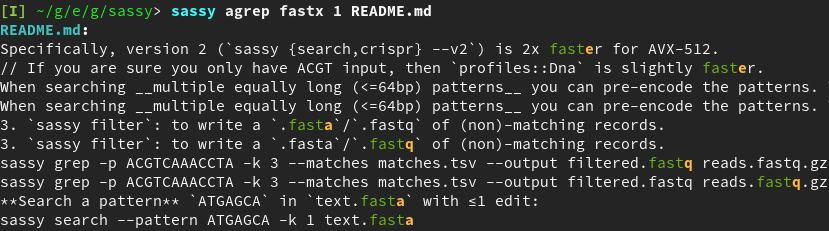
Figure 1: Output of sassy agrep fastx 1 README.md.
For DNA, the common usage is sassy grep, which can look like this:
| |
If you have a file with many patterns, instead use --pattern-fasta or --pattern-file:
| |
Add the --v2 flag to enable version 2, which is significantly faster when all
patterns have the same length.
The output of sassy grep and the .tsv of sassy search contain a number of
fields:
| |
- the pattern ID (when searching multiple patterns);
- the ID of the matching record;
- the number of errors in the match;
- the strand:
+for forward matches, or-when the text had to be reverse-complemented first; - the 0-based start and exclusive end position in the forward direction (as in the input file);
- the extracted matching region, possibly after reverse complementing it for
-matches. Use--samto avoid this and be compatible with the SAM spec. - The CIGAR string between the pattern and the matching region.
Dis a character in the pattern that was deleted in the text andIis in the text but not the pattern. The traceback prefers matches and mismatches over insertions and deletions.
2.2 Rust library Link to heading
Sassy also comes as a Rust crate (docs). The basic API is:
| |
The Sassy2 API for searching many equal-length patterns uses encode_patterns.
When applicable, this is the fastest version of Sassy.
| |
When searching one or more variable-size patterns across one or more texts,
search_many provides a single API endpoint that handles the multi-threading and
batching. It supports multiple ways of filling the SIMD lanes: with multiple
chunks of a single text (the Sassy1 default), with chunks from multiple
(similar-length) texts, or with chunks from multiple (similar-length) patterns.
| |
2.3 Python bindings Link to heading
This is a simple as pip install sassy-rs, followed by
| |
To search many patterns across many texts at once without going through the API multiple times, use
| |
For more details, see the python bindings readme.
2.4 C and R bindings Link to heading
See the C bindings readme and the third-party Rsassy package.
3 Problem statement: Approximate String Matching Link to heading
As already mentioned, Sassy finds all matches of your pattern. While this may seem natural, this sets it apart from most other tools that only return a single best match. In particular, many alignment libraries such as parasail (Daily 2016), Seqan (Döring et al. 2008), and Edlib (Šošić and Šikić 2017) are primarily designed to only return a single best semi-global alignment with a minimal number of edits. But if the data is contaminated, this could occur anywhere, and a pattern could occur multiple times in a read.
Thus, Navarro (2001) defines the problem as follows:
Given a pattern \(P\) of length \(m\) and a text \(T\) of length \(n\), find all end positions \(j\in \{0,\dots n\}\) in the text for which there exists a start position \(0\leq i\leq j\) such that the pattern matches \(T[i\dots j)\) with at most \(k\) errors.
There are a few things to note:
- ASM only asks for the end positions. Not full alignments, nor start positions of the alignments. By default, indeed only endpoints are returned, but if requested, Sassy will report a single alignment per endpoint2.
- The definition above asks for all end positions, but this is not quite practical:
In the figure below, which shows the edit graph, we see that there are three
consecutive positions (in the bottom row) where the pattern matches with 1
error. But the corresponding alignments overlap, and so reporting all of them
makes little sense. Thus, by default, Sassy only reports the rightmost match in every
local minimum. The
search_allAPI does return all positions. - While not explicitly stated, Sassy supports IUPAC characters, so that an
Nmatches with all other bases.

Figure 2: The edit graph (see my thesis chapter for an introduction) of aligning pattern ABB to text ABAB. The grey cells on the top and left indicate the initialization, and the computed output values in the bottom row are shaded yellow. The optimal alignments paths are highlighted in yellow as well.
Another thing to note here is that ASM is not invariant under reversing the input strings: we see on the right (where we fill the matrix from bottom-right to top-left) that now there are two local minima rather than just one before.
3.1 Overhang: when the pattern extends beyond the read Link to heading
Reads can be short, which means that half of an adapter might be cut off at the
end of a read. To support searching these as well, Sassy implements a special
overhang mode where overhanging (cut-off) characters get a penalty of \(\alpha < 1\),
which defaults to \(\alpha = 0.5\). In the example below, this means there are
overhanging matches of ACBBA on both the left and right end of the text, both
for cost 1.

Figure 3: Sassy supports cases where the pattern extends beyond the text/read, as indicated by the yellow alignment on the left and right. Here, these only have cost 1 instead of 2 and 3 respectively.
4 Some background Link to heading
As a small aside, you might be familiar with my previous work on A*PA (Groot Koerkamp and Ivanov 2024) and A*PA2 (Groot Koerkamp 2024) (GitHub), which is a global aligner instead that runs in near-linear time when the two input sequences are similar. They work particularly well for sequences up to 1 Mbp. Unfortunately, it turns out that such alignments are rare in practice since structural variation breaks such long-range correlations.
The conclusion is basically that doing a lot of dumb work very fast is often better than being clever about it. Thus, in Sassy we do away with all the fancy stuff from A*PA[2] and leave just the fast DP kernel.
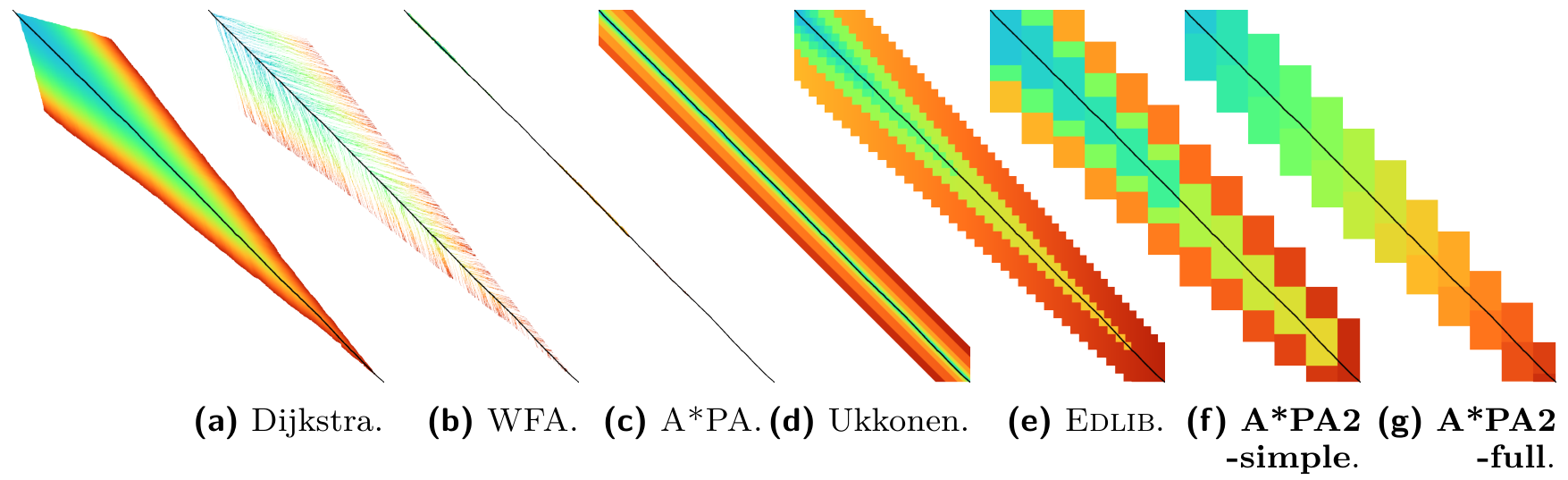
Figure 4: Figure from the A*PA2 paper showing the states computed by various algorithms. A*PA computes only very few states along the main diagonal, but comes with a large overhead. A*PA2 processes states in big (256times 256) blocks, but ends up being faster.
5 The algorithm Link to heading
Algorithmically, Sassy does not do much new, but nevertheless, combining a number of existing techniques and implementing them efficiently gives good results.
If you’re familiar with the Needleman-Wunsch DP algorithm for edit distance and the corresponding edit graph, then the basic algorithm should be familiar. (Otherwise, see my thesis chapter for an introduction to alignment concepts and algorithms.)
The naive approach is to simply fill the entire \(n\times m\) DP matrix in \(O(nm)\) time, but this can be sped up in various ways. First, Myers’ bitpacking algorithm (Myers 1999) provides a clever way of using bit-tricks to compute 64 cells of the DP matrix at once. Whereas Myers' uses “vertical” blocks, splitting the pattern into chunks of size \(k\), we use horizontal blocks, splitting the text into chunks of size \(k\), as shown in the figure below. The main benefit here is that often, the pattern length \(m\) is less than 64, so that tiling them vertically would waste most of the bits.
Similar to Myers’ method, we can also use an “early break” scheme: we intend to find all states of the DP matrix that have a cost \(\leq k\). This means that as soon as all states in a row are at distance \(>k\), no good alignment can go through them, and we can stop there. In practice, the cost of aligning two random DNA sequences is around 45% of their length, meaning that the cost of the alignment will exceed \(k\) after roughly \(2k\) rows when two sequences are unrelated.
Together, this means that on random text (without matches), Sassy only computes only around \(n\cdot 2k = O(nk)\) DP states, and it does so \(w=64\) at a time, for an overall complexity of \(O(\lceil n/w\rceil k)\).

Figure 5: An example of the states of the DP matrix computed by Sassy. The highlighted region shows states at distance (leq k). Horizontal blocks of 64 states are computed all at once. The early break stops processing a column as soon as all states in it are at distance (>k). In this example, there is one match where an alignment of cost (leq k) makes it to the bottom row.
5.1 SIMD Link to heading
We use SIMD instructions to significantly speed up the algorithm. Instead of computing one column at a time, AVX2 provides 256-bit registers that can fit 4 64-bit values. Thus, we split the text into 4 chunks and then process these at the same time. A SIMD register then contains one block from each of the 4 chunks, as highlighted in blue.
We also tried using AVX-512, but that this not provide additional speedups. The problem there is that reading from 8 chunks of text in parallel and “transposing” this data to the right SIMD lanes becomes a bottleneck.

Figure 6: Sassy chunks the text into 4 chunks and uses SIMD instructions to process them in parallel.
5.2 Sassy2 Link to heading
The algorithm so far was designed for searching a single pattern, which is great for a CLI. But it turns out that in practice, users like to search many short and equal-length patterns at once. For example, the 96 demultiplexing barcodes, or many candidate crisper primers.
Rick Beeloo designed Sassy2 (Beeloo and Groot Koerkamp 2026a) (PDF, DOI) for this case specifically, and it makes a number of different choices. Rather than filling SIMD lanes with text chunks, we now use batching of patterns and each lane is used to search a separate pattern. Furthermore, we can now switch back to vertical tiling, but instead of using full 64-bit lanes, we use smaller \(w’\)-bit lanes: 8 bits for exact matching, 16 bits when \(1\leq k < 4\), 32 bits when \(4\leq k<8\), and 64 bits otherwise. For small \(k=3\), this allows packing 16 patterns in a 256-bit AVX2 register, and even 32 patterns in a 512-bit AVX512 register!
Thus, we take the suffix of length \(w’\) of each pattern and find where this matches with \(\leq k\) errors, as highlighted in yellow in the figure below. These locations mark the possible endpoints of full matches, and so these candidate regions are verified in a second stage.

Figure 7: Sassy2 takes a suffix of length (w’) of each pattern and aligns (L’ = 256/w’) (or (L’=512/w’)) patterns at once by using a SIMD register (left blue column). Locations where a pattern matches with cost (leq k) are verified in a second stage, shown on the right.
6 Results Link to heading
6.1 Sassy1 Link to heading
This first plot is for searching a long text (\(n=10^5\)) in the forward-only direction using a single thread, with patterns of varying length and varying \(k\). We see that with \(k=3\), Sassy1 reaches up to 1.1 GB/s, and still has 600 MB/s for \(k=20\). Sassy is up to 10\(\times\) faster than Edlib and 128\(\times\) faster than Parasail.

Figure 8: Log-log plot of the search throughput of Parasail, Edlib, and Sassy for varying pattern lengths.
If we fix the pattern length at \(m=23\) and instead vary the length of the text, we see that sassy only reaches its maximum throughput for texts of length at least 4000 characters or so. The main reason for the relatively inefficient search at short sequences is that splitting the text into 4 chunks requires the chunks to be aligned to 64-character boundaries and also requires some overlap between adjacent chunks, which together cause inefficiencies when \(n\) is on the order of the 256-bit SIMD-width.

Figure 9: Log-log plot of the search throughput of Parasail, Edlib, and Sassy for varying text lengths.
6.2 Sassy2 Link to heading
When searching many many short and equal-length patterns, Sassy2 significantly improves the throughput of Sassy1, going up to 8 Gbp/s, and also it does not slow down when searching short texts.
Reasons that Sassy2 outperforms Sassy1 are that the early-break check in each column in v1 is somewhat expensive, and that the vertical tiling allows for a more efficient SIMD implementation of determining which pattern characters match the text.

Figure 10: Log-log plots of the search throughput of Sassy2 for searching a pattern of length (m=23) on 1 thread and varying (kin{0,3,4}) (with (w’={8,16,32}) respectively). A: searching 128 patterns in a text of varying length. B: searching increasingly many patterns with (k=3) errors in a (n=10^5) long text.
On the right, we see a linear speedup from increasing the number of patterns to 16 (AVX2) and 32 (AVX-512), since this simply fills the additional available SIMD lanes. On the left, we see that the throughput only goes down slightly for shorter texts, from 8 Gbp/s for \(n=8000\) to 5 Gbp/s for \(n=150\). At that point, Sassy2 is nearly 256x as fast as Edlib.
6.3 Applications Link to heading
As for applications, Sassy1 is great for one-off searches as done by the CLI, while Sassy2 shines when searching batches of patterns.
For Crispr off-target detection, searching many 23bp guide RNAs in a human genome with \(k=3\) takes only 30ms per gRNA, totalling >100 Gbp/s search throughput on 16 threads. That’s 3.7\(×\) faster than Sassy1 and 35\(×\) faster than Edlib.
Scanning Nanopore reads for 96 ONT rapid barcodes with \(k=3\) runs at 1.2 Gbp/s, 4.6\(×\) Sassy1, and 45\(×\) Edlib. This forms the basis of Barbell (Beeloo et al. 2026) (DOI), a fast and accurate demultiplexer also developed by Rick Beeloo. Specifically, Barbell uses Sassy2 to quickly scan ONT reads to detect and trim barcodes and adapters.
Discuss on reddit.